
Google DeepMind: Millions of new materials discovered with deep learning
"With GNoME, we’ve multiplied the number of technologically viable materials known to humanity."
Developing new materials is crucial for technological progress yet remains a huge challenge. Materials underpin innovations across industries from clean energy to information processing. Novel materials have enabled fundamental breakthroughs like lithium-ion batteries, high-temperature superconductors, LEDs, and more.
However, the space of possible materials combinations is enormous. Traditional experimental approaches involve synthesizing and testing thousands of different compositions through trial and error. This process is prohibitively slow, expensive, and labor-intensive given the vast number of potential candidates. Computational screening offers a way to dramatically accelerate discovery by evaluating candidates efficiently in silico before experiments. But historically, machine learning struggled to accurately model material properties like stability for large-scale screening.
Overcoming these barriers could revolutionize how new materials are discovered. Enabling rapid computational discovery of novel functional materials would fuel innovation across industries and tackle grand challenges in areas like energy storage. In this post, we'll take a look at how researchers at Google DeepMind built a tool called GNoME that tackles this challenge head-on. Let's begin!
Technical Description of the GNoME Framework
Before we get any further, let's first turn to the project's announcement page on DeepMind to get some additional context:
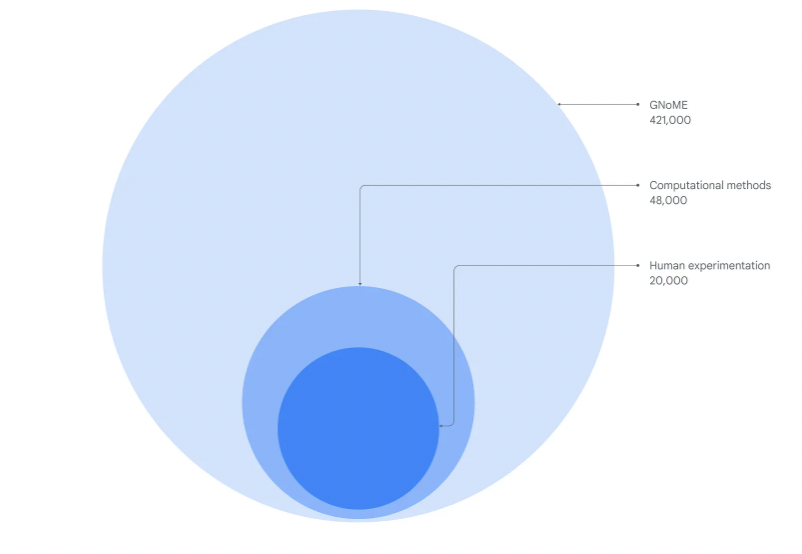
In the past, scientists searched for novel crystal structures by tweaking known crystals or experimenting with new combinations of elements - an expensive, trial-and-error process that could take months to deliver even limited results. Over the last decade, computational approaches led by the Materials Project and other groups have helped discover 28,000 new materials. But up until now, new AI-guided approaches hit a fundamental limit in their ability to accurately predict materials that could be experimentally viable. GNoME’s discovery of 2.2 million materials would be equivalent to about 800 years’ worth of knowledge and demonstrates an unprecedented scale and level of accuracy in predictions.
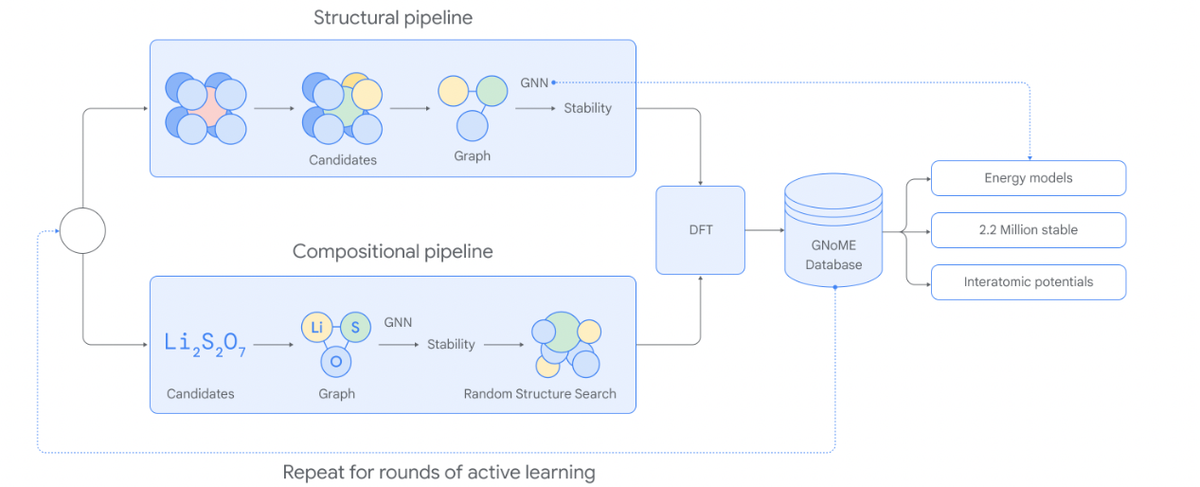
The researchers developed Graph Networks for Materials Exploration (GNoME), a novel framework that combines graph neural networks with an active learning approach to scale up computational materials discovery.
Graph neural networks (GNNs) are a type of neural network well-suited for modeling structured, relational data-like materials. GNoME models take in a material's crystal structure encoded as a graph, with atoms as nodes and bonds as edges. The model passes messages between connected atoms to update representations, capturing the local environment and long-range interactions.
An initial GNoME model was trained on around existing materials (check out the Materials Project for more on this) to predict formation energies. GNoME then iteratively generates candidate materials through substitution and structural perturbations. Candidates are filtered by the model, factoring in uncertainties from ensemble predictions.
The energy of filtered candidates is evaluated using density functional theory (DFT) calculations, the most accurate computationally feasible technique. These results revalidate model predictions while expanding the training set. Models are retrained on this augmented data in each round, driving improvement. This active learning loop enables efficiently scaling up materials discovery through a self-supervised "data flywheel" effect.
To maximize candidate diversity beyond substitutions, GNoME introduced Symmetry-Aware Partial Substitutions (SAPS). SAPS generalizes common substitution frameworks to enable incomplete replacements respecting crystal symmetries. This discovers novel derivative structures while exploiting common prototypes.
How the GNoME Models Were Evaluated
The researchers extensively evaluated GNoME across multiple rounds of active learning and model scaling. Key metrics are available in the paper. But what's really interesting is that predicted structures were validated in the real world by labs!
Over 2.2 million previously unknown stable crystal structures were discovered, expanding the known frontier by nearly an order of magnitude. Of these, "380,000 are the most stable, making them promising candidates for experimental synthesis. Among these candidates are materials that have the potential to develop future transformative technologies ranging from superconductors, powering supercomputers, and next-generation batteries to boost the efficiency of electric vehicles."
736 structures have since been experimentally realized independently, in the lab, by external researchers. Analyses found most discoveries indeed stably differed from known phases, and the majority remained stable.
The impact of scaling behavior was analyzed, finding GNoME predictions improved as a power law with data size - suggesting continuing breakthroughs are possible. Overall, these results demonstrate the transformative potential of scaling up deep learning for materials discovery.
Realizing New Materials Modelling Capabilities
In addition to novel discoveries, GNoME's extensive dataset unlocked new modeling capabilities through machine learning. The relaxed trajectories presented an unparalleled range of atomic configurations to train interatomic potentials - models that describe interactions between atoms.
A graph network potential pre-trained on GNoME exhibited unprecedented zero-shot accuracy without any materials-specific training. It enabled molecular dynamics simulations at atomic resolution for screening new solid electrolytes, discovering promising candidates computationally that experiments could now target.
This work shows scaling machine learning through access to large and diverse materials data unleashes modeling abilities beyond what was previously conceived. Powerful pre-trained models may accelerate discovery across many applications by providing accurate yet general physics-based simulations out of the box.
Conclusion and Broader Impacts
Overall, this research demonstrates the immense potential of scaling up deep learning to transform how new materials are discovered. Their efforts expanded the frontiers of known stable materials by nearly an order of magnitude through computational screening. These methodologies now enable exploring regions of composition space previously inaccessible.
The profusion of novel discoveries could fuel unprecedented innovation if experimentally realized. New insights into material behaviors are also empowering further computational discovery. As modeling and data-driven tools continue advancing in lockstep, our ability to address major societal challenges through engineered functionality at the nanoscale will be greatly accelerated. Following in GNoME's footsteps continued joint progress on machine learning and scientific discovery could have transformative impacts.

Comments ()